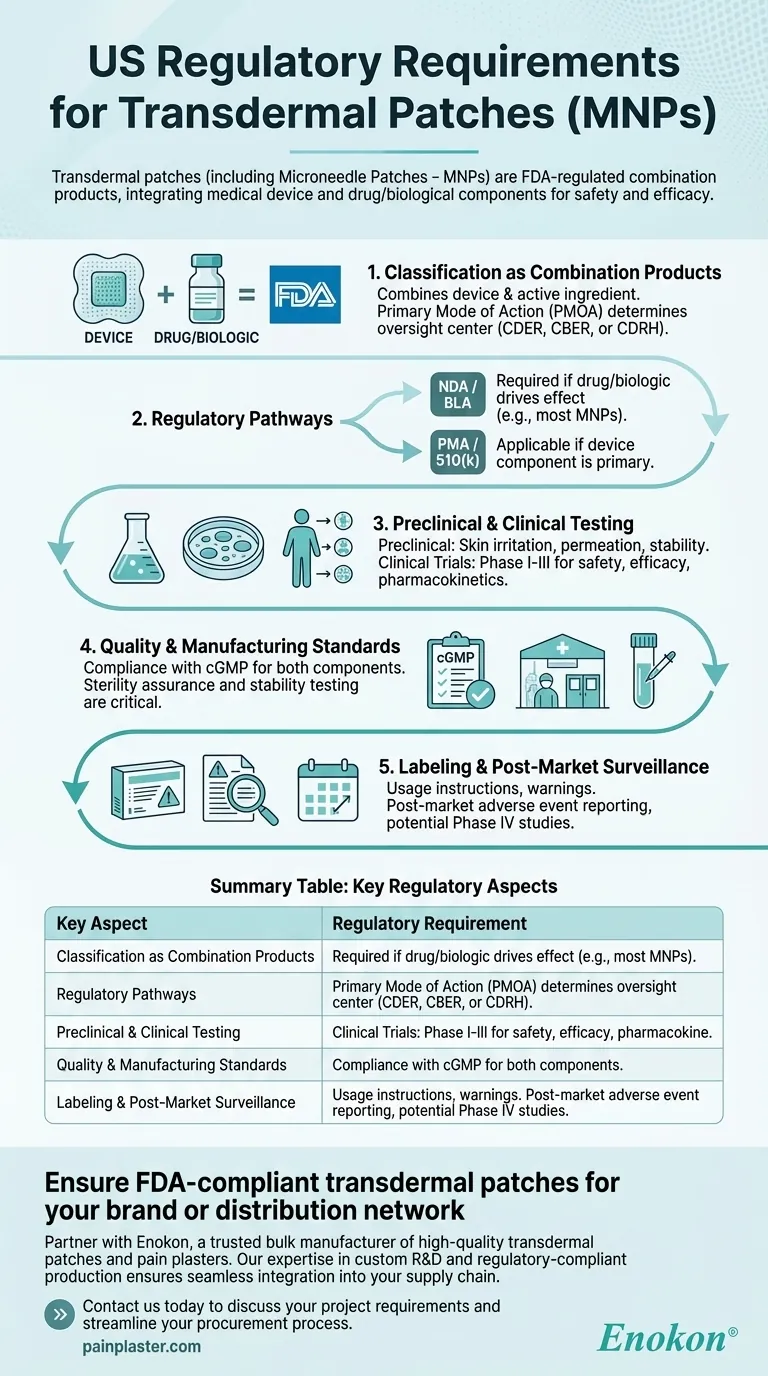
마이크로니들 패치(MNP)를 포함한 경피 패치는 미국에서 FDA에서 복합 제품으로 규제하고 있으며, 안전성과 효능을 보장하기 위해 엄격한 승인 절차를 거쳐야 합니다. 이러한 제품은 의료 기기와 약물/생물학적 성분이 통합되어 있어 주요 작용 방식에 따라 특정 규제 경로를 준수해야 합니다. 승인 절차에는 전임상 및 임상 테스트, 품질 관리, 라벨링 및 제조 표준 준수 등이 포함됩니다.
핵심 사항을 설명합니다:
-
복합 제품 분류
- FDA는 다음과 같이 분류합니다. 경피 패치 는 의료 기기(예: 패치 백킹, 마이크로니들)와 약물 또는 생물학적 활성 성분을 결합하기 때문에 복합 제품으로 분류합니다.
- 주 작용 방식(PMOA)에 따라 해당 제품이 약물평가연구센터(CDER), 생물의약품평가연구센터(CBER) 또는 의료기기 및 방사선보건센터(CDRH)의 규제를 받는지 여부가 결정됩니다.
-
규제 경로
- 신약 신청(NDA) 또는 생물학적 제제 허가 신청(BLA): 약물 또는 생물학적 성분이 치료 효과를 유발하는 경우 필요합니다.
- 시판 전 승인(PMA) 또는 510(k): 기기 구성 요소가 주요한 경우 적용 가능(예: 약물 전달을 촉진하는 마이크로니들).
- MNP는 약물 중심적 기능으로 인해 NDA/BLA 경로를 따르는 경우가 많습니다.
-
전임상 및 임상 시험
- 전임상 연구: 피부 자극, 투과성 및 안정성 테스트를 포함하여 안전성과 전달 효율성을 평가합니다.
- 임상 시험: 1상~8상 임상시험은 사람을 대상으로 약동학, 효능 및 부작용을 평가합니다.
-
품질 및 제조 표준
- 약물 및 기기 구성 요소 모두에 대해 현재 우수 제조 관리 기준(cGMP)을 준수합니다.
- 생물학적 제제가 포함된 패치의 경우 무균성 보증 및 안정성 테스트가 중요합니다.
-
라벨링 및 시판 후 감시
- 라벨에는 사용 지침, 경고 및 보관 조건이 포함되어야 합니다.
- 시판 후 요건에는 부작용 보고 및 잠재적인 4상 임상시험이 포함됩니다.
구매자의 경우 이러한 요건을 이해하면 FDA 규정을 준수하는 공급업체와 연계하여 부적합 제품의 위험을 줄일 수 있습니다. 이러한 규정이 조달 일정이나 공급업체 선정 기준에 어떤 영향을 미치는지 고려해 보셨나요? 장치와 약물 감독의 상호 작용은 최신 경피 치료의 신뢰성을 조용히 형성합니다.
요약 표:
| 주요 측면 | 규제 요건 |
|---|---|
| 분류 | FDA에서 복합 제품(기기 + 약물/생물학적 제제)으로 규제합니다. |
| 주요 조치 방식 | CDER(약물), CBER(생물학적제제) 또는 CDRH(기기)에 의해 감독을 결정합니다. |
| 승인 경로 | NDA/BLA(약물 중심) 또는 PMA/510(k)(기기 중심). MNP는 일반적으로 NDA/BLA를 따릅니다. |
| 테스트 요구 사항 | 전임상(안전성, 투과성) 및 임상시험(1~3상). |
| 제조 표준 | 약물 및 기기 구성 요소 모두에 대한 cGMP 준수, 생물학적 제제에 대한 무균 보증. |
| 시판 후 의무 | 부작용 보고, 4상 임상시험 및 라벨링 규정 준수. |
브랜드 또는 유통 네트워크에 대한 FDA 준수 경피 패치 보장
파트너
Enokon
고품질 경피 패치 및 통증 플라스터의 신뢰할 수 있는 대량 제조업체입니다. 맞춤형 R&D 및 규정 준수 생산에 대한 당사의 전문 지식은 귀사의 공급망에 원활하게 통합될 수 있도록 보장합니다.
지금 바로 문의
에 문의하여 프로젝트 요구 사항을 논의하고 조달 프로세스를 간소화하세요.
시각적 가이드
관련 제품
- 멘톨 젤 통증 완화 패치
- 아이시 핫 멘톨 약용 통증 완화 패치
- 생리통을 위한 온열 통증 완화 패치
- 성인과 어린이를 위한 천식 기침 및 통증 완화 패치
- 소화기 완화를 위한 허브 약용 설사 방지 패치